即将执行8.2化妆品中α-熊果苷等4种原料的检验方法 的CP柱数据分享
发布时间:
近日,国家药品监督管理局发布了“国家药监局关于将油包水类化妆品的pH值测定方法等21项制修订项目纳入化妆品安全技术规范(2015年版)的通告(2023年第41号)”。
![]()
对《化妆品安全技术规范(2015年版)》中原有的5项检验方法进行了修订,自2024年3月1日起,化妆品注册、备案及抽样检验相关检验应当采用新发布的检验方法。
新增14项检验方法,纳入《化妆品安全技术规范(2015年版)》,自通告发布之日起实施。
今天给大家带来的是“化妆品中α-熊果苷等4种原料的检验方法”的数据分享。
使用CAPCELL PAK MGIII C18色谱柱可以得到α-熊果苷、β-熊果苷相对较好的分离,目标峰线性、回收率良好。
使用CAPCELL PAK ADME-HR色谱柱,对色谱条件做一些调整后,方法验证可得到良好分析结果,线性、回收率良好,同时α-熊果苷和β-熊果苷的分离度可达到3.58,并缩短了整体分析时间。
01
- 不同保留机理色谱柱的比较
首先使用不同保留机理的色谱柱对α-熊果苷和β-熊果苷的分离效果进行比较。
在常规C18体系下,CAPCELL PAK C18 MGIII可以得到较好的分离效果,而使用键合金刚烷基团的CAPCELL PAK ADME-HR(金刚烷键合)色谱柱,在相同液相条件下能得到更好的分离效果,分析结果如图1所示。
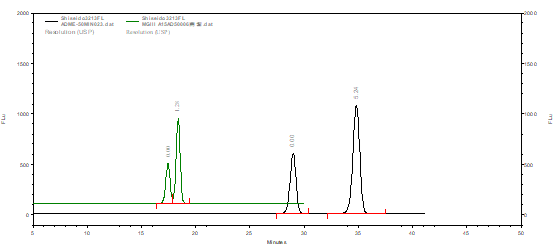
图1 C18 MGIII和ADME-HR色谱柱
α-熊果苷、β-熊果苷对照品分析结果对比图
【色谱条件】
色谱柱:CAPCELL PAK C18 MGIII S5;4.6×250
CAPCELL PAK ADME-HR S5;4.6×250
流动相:水 / 甲醇 = 95 / 5
流速:0.5 mL/min
柱温:20 ℃
检测:激发波长:292;发射波长:337
样品:α-熊果苷20μg/mL,β-熊果苷10μg/mL (溶剂为甲醇)
进样量:5 μL
02
参考检验方法得到的结果
参考检验方法中推荐的色谱柱类型和规格,使用CAPCELL PAK C18 MGIII色谱柱对标准品、乳液样品和化妆水样品进行分析,可获得良好的分析结果。
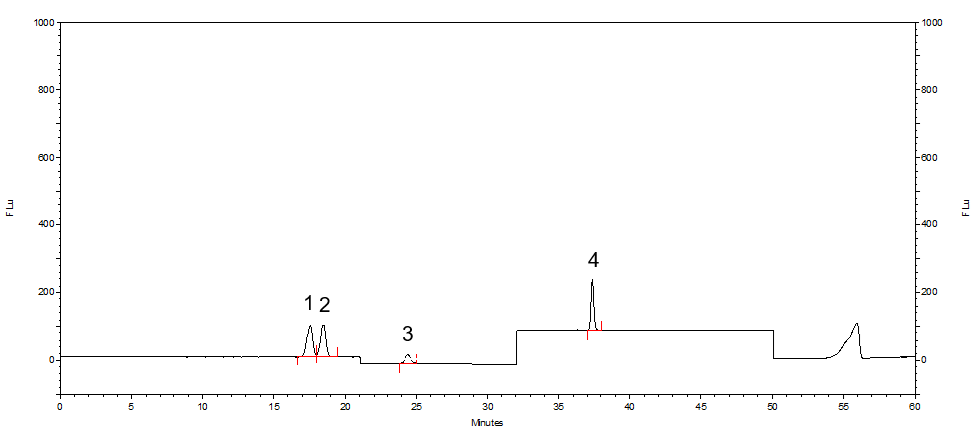
图2 MGIII色谱柱分析结果-标准溶液
1.β-熊果苷 2.α-熊果苷 3.氢醌 4.苯酚
表1 MGIII色谱柱标准溶液中各峰积分结果表
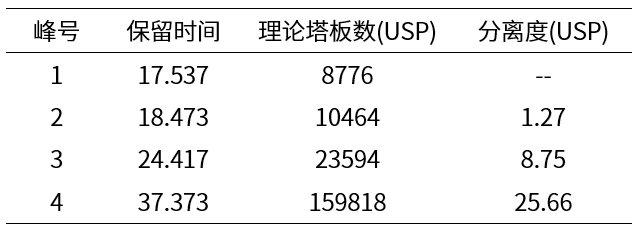
【色谱条件】
色谱柱: CAPCELL PAK MGIII C18, 4.6 mm×250 mm, 5 μm(A15AD 50006)
流动相:A:水 B:甲醇
B% 5%(0min)-5%(21min)->45%(22min)->50%(43min)->50%(48min)->100%(48.1min)->100%(50min)->5%(50.1min)->5%(60min)
流速:0.5 mL / min
柱温:20 ℃
进样量: 5μL
检测:α-熊果苷、β-熊果苷的激发波长为292,发射波长为337;
氢醌的激发波长为298,发射波长为345;
苯酚的激发波长为:280,发射波长为318
样品:α-熊果苷2μg/mL,β-熊果苷2μg/mL,氢醌0.2μg/mL,苯酚0.4μg/mL (溶剂为甲醇)
标准曲线
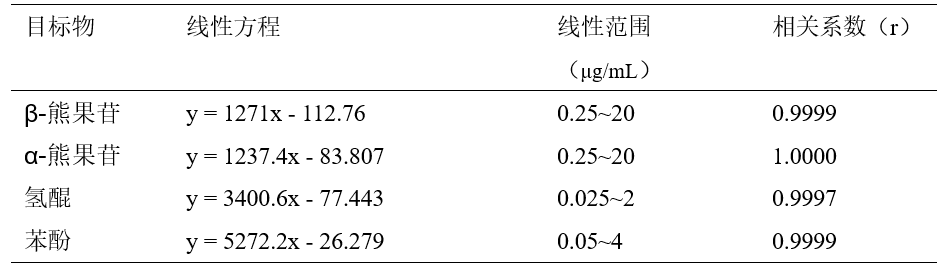
用甲醇将固体标准品溶解稀释配制成标准工作溶液,浓度依次由低向高进样测定,以峰面积-浓度作图,绘制标准工作曲线。标准曲线在浓度范围内线性良好,线性系数r的范围为0.9997~1.0000,测试结果如表2所示。
表2 α-熊果苷、β-熊果苷、氢醌和苯酚的线性方程、线性范围和相关系数
回收率
按照标准方法分别对化妆水和乳液样品进行加标回收实验,α-熊果苷和β-熊果苷添加量为20μg,氢醌添加量为2μg,苯酚添加量为4μg。对样品和加标样品进行分析,计算得到回收率结果,分析谱图如图3和图4所示。化妆水样品和乳液样品加标回收率范围为91.3%~108.9%,回收率良好。
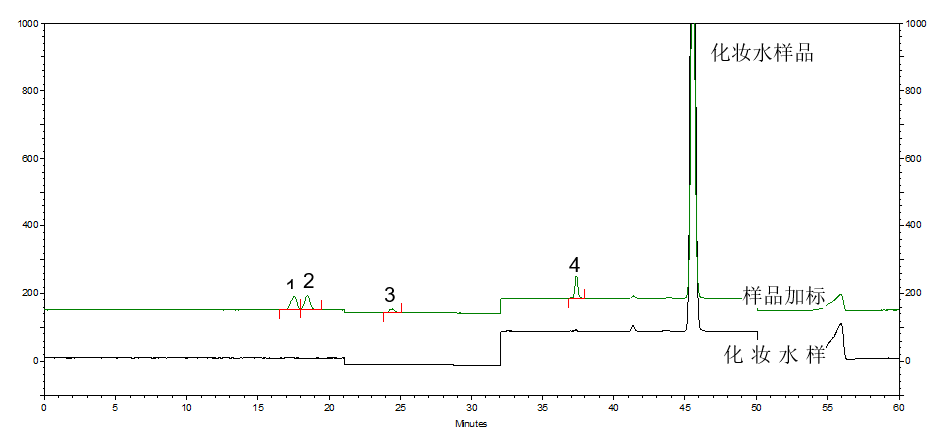
图3 化妆水样品和化妆水加标样品分析图
1.β-熊果苷 2.α-熊果苷 3.氢醌 4.苯酚
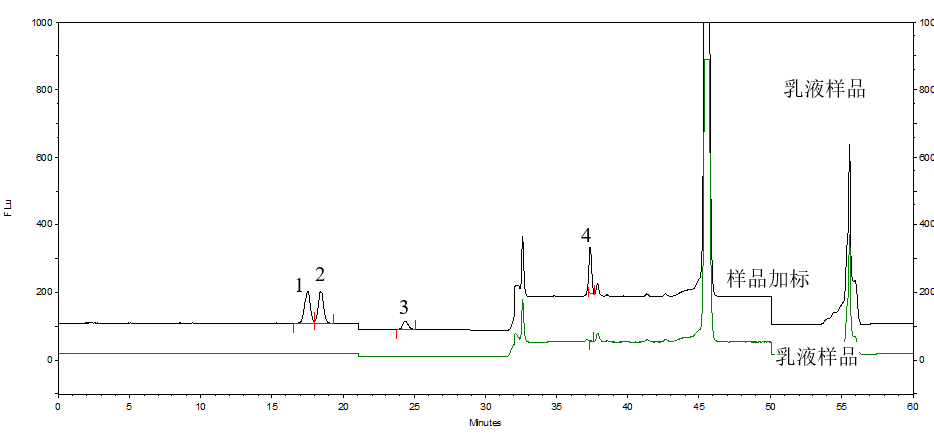
图4 乳液样品和乳液加标样品分析图(样品中苯酚有值需要质谱确认)
1.β-熊果苷 2.α-熊果苷 3.氢醌 4.苯酚
【色谱条件】
色谱柱: CAPCELL PAK MGIII C18, 4.6 mm×250 mm, 5 μm(A15AD 50006)
流动相: A:水 B:甲醇
B%5%(0min)->5%(21min)->45%(22min)->50%(43min)->50%(48min)->100%(48.1min)->100%(50min)->5%(50.1min)->5%(60min)
流 速: 0.5 mL / min
柱 温: 20°C
进样量: 5μL
检 测: α-熊果苷、β-熊果苷的激发波长为292,发射波长为337;氢醌的激发波长为298,发射波长为345;苯酚的激发波长为280,发射波长为318
样 品: 称取样品1.0g(精确到0.001g),置10mL具塞比色管中,加入甲醇至10mL,在涡旋混匀器上高速振荡30s,使样品与提取溶剂充分混合。密塞,超声提取20min,静置至室温,取适量样品溶液离心(转速14000r/min)5min,取上清液经0.45μm微孔滤膜过滤,取续滤液作为待测溶液。必要时用适量甲醇稀释。
03
使用CP ADME-HR的结果
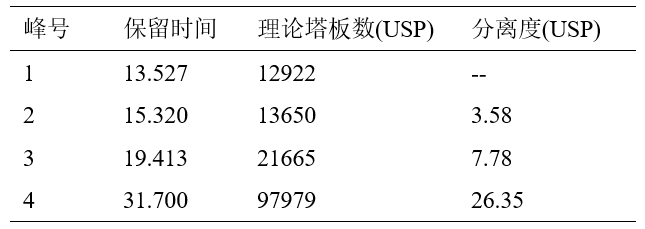
基于α-熊果苷和β-熊果苷差向异构的化学性质,使用键合金刚烷基团的CAPCELL PAK ADME-HR色谱柱,在检验方法的液相条件下能得到更好的分离效果,但保留时间较长,和氢醌峰接近,所以检验方法中的梯度条件不适合此款色谱柱,我们尝试在检验方法的基础上对色谱条件做一些调整,使目标峰能够获得更好的分离。通过相应的方法学验证后,发现新方法下线性、回收率良好,同时α-熊果苷和β-熊果苷的分离度可达到3.58,并缩短了测试时间。
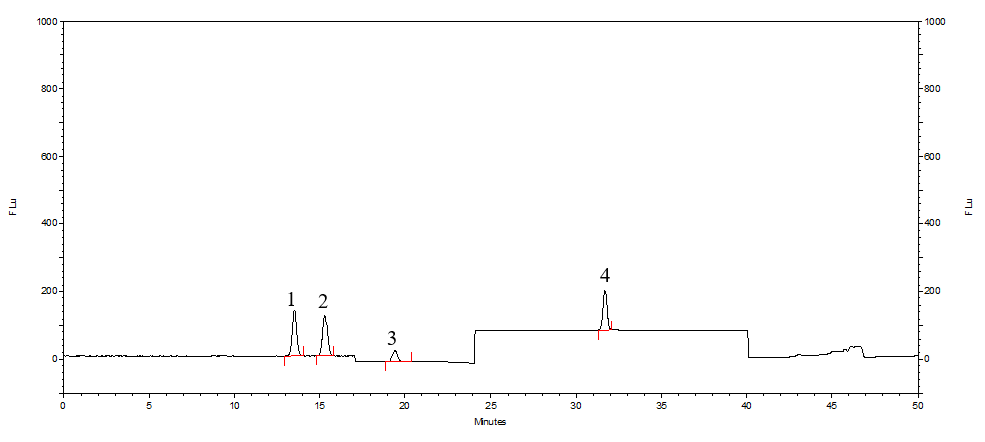
图5 ADME色谱柱分析结果-标准溶液
1.β-熊果苷 2.α-熊果苷 3.氢醌 4.苯酚
表3 ADME色谱柱标准溶液中各峰积分结果表
【色谱条件】
色谱柱: CAPCELL PAK ADME HR, 4.6 mm×250 mm, 5 μm(V45HAD01279)
流动相: A:水 B:甲醇
B%13%(0min)->13%(15min)->45%(15.1min)->49%(35min)->49%(36min)->100%(36.1min)->100%(40min)->13%(40.1min)->13%(50min)
流 速: 0.5 mL / min
柱 温: 30°C
进样量: 5μL
检 测: α-熊果苷、β-熊果苷的激发波长为292,发射波长为337;氢醌的激发波长为298,发射波长为345;苯酚的激发波长为280,发射波长为318
样 品: α-熊果苷2μg/mL ;β-熊果苷2μg/mL;氢醌0.2μg/mL;苯酚0.4μg/mL (溶剂为甲醇)
标准曲线
用甲醇将固体标准品溶解稀释配制成标准工作溶液,浓度依次由低向高进样测定,以峰面积-浓度作图,绘制标准工作曲线。标准曲线在浓度范围内线性良好,线性系数r的范围为0.9998~0.9999。测试结果如表4所示。
表4 α-熊果苷、β-熊果苷、氢醌和苯酚的线性方程、线性范围和相关系数
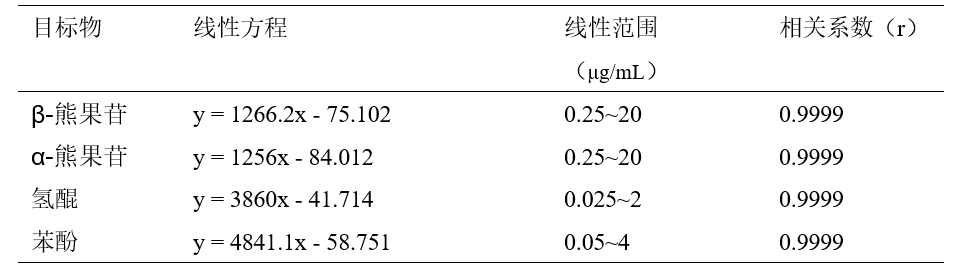
回收率
按照标准方法分别对化妆水和乳液样品进行加标回收实验,α-熊果苷和β-熊果苷添加量为20μg,氢醌添加量为2μg,苯酚添加量为4μg。对样品和加标样品进行分析,计算得到回收率结果,分析谱图如图6和图7所示。化妆水样品和乳液样品加标回收率范围为88.4%~99.9%,回收率较好。
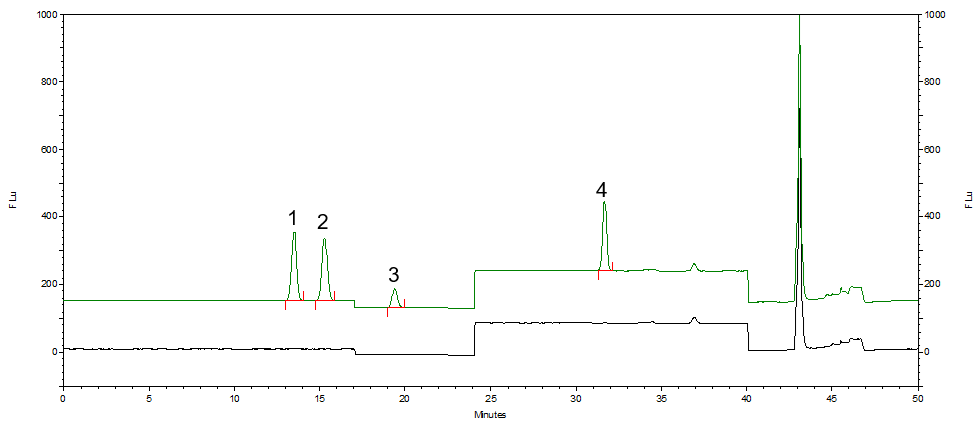
图6 化妆水样品和化妆水加标样品分析图
1.β-熊果苷 2.α-熊果苷 3.氢醌 4.苯酚
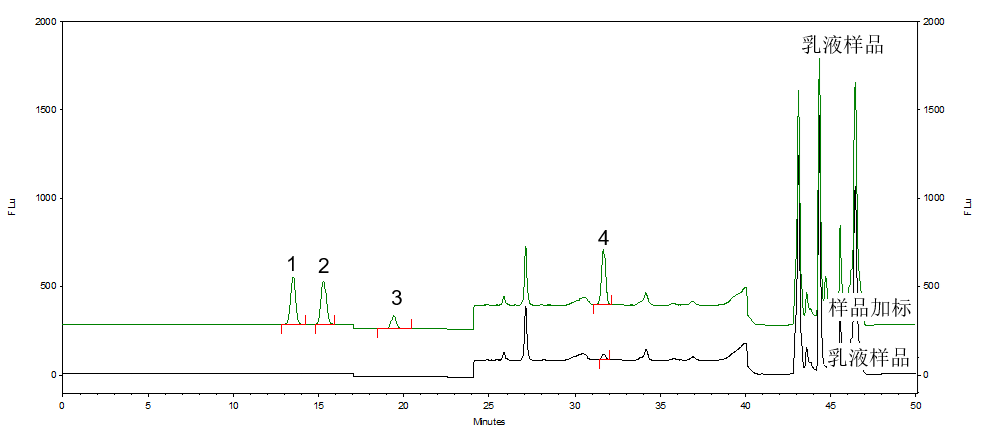
图7 乳液样品和乳液加标样品分析图(样品中苯酚有值需要质谱确认)
1.β-熊果苷 2.α-熊果苷 3.氢醌 4.苯酚
【色谱条件】
色谱柱: CAPCELL PAK ADME HR, 4.6 mm×250 mm, 5 μm(V45HAD01279)
流动相: A:水 B:甲醇
B%13%(0min)->13%(15min)->45%(15.1min)->49%(35min)->49%(36min)->100%(36.1min)->100%(40min)->13%(40.1min)->13%(50min)
流 速: 0.5 mL / min
柱 温: 30°C
进样量: 5μL
检 测: α-熊果苷、β-熊果苷的激发波长为292,发射波长为337;氢醌的激发波长为298,发射波长为345;苯酚的激发波长为280,发射波长为318
样 品: 称取样品1.0g(精确到0.001g),置10mL具塞比色管中,加入甲醇至10mL,在涡旋混匀器上高速振荡30s,使样品与提取溶剂充分混合。密塞,超声提取20min,静置至室温,取适量样品溶液离心(转速14000r/min)5min,取上清液经0.45μm微孔滤膜过滤,取续滤液作为待测溶液。必要时用适量甲醇稀释。
结论
综上,参考《化妆品安全技术规范 (2015年版)》第四章理化检验方法中8.2化妆品中α-熊果苷等4种原料的检验方法,使用CAPCELL PAK C18 MGIII S5;4.6×250色谱柱可以得到α-熊果苷和β-熊果苷较好的分离,目标峰线性和回收率良好。
使用CAPCELL PAK ADME-HR S5;4.6×250色谱柱,对检验方法进行调整后,可得到良好分析结果,线性和回收率良好,同时α-熊果苷和β-熊果苷的分离度可达到3.58,并缩短了测试时间。